


A continuación encontraréis unos consejos muy útiles sobre el pku, con ello queremos conseguir que las vida y sus familias sean lo más cómodas posible dentro del sacrificio que conlleva una dieta de por vida.
Información sobre la enfermedad
Fenilcetonuria-PKU
La Fenilcetonuria es una enfermedad hereditaria que consiste en una alteración del metabolismo de los aminoácidos, y si no se trata desde los primeros días de vida del recién nacido , impedirá el normal desarrollo del niño/a, llegando incluso a producirle un retraso mental profundo.
¿Cómo sabré si mi hijo tiene una Fenilcetonuria(P.K.U.)?
Existe una prueba para detectar de manera precoz esta enfermedad, que popularmente se conoce como "la prueba del talón".
La Conselleria de Sanidad realiza la prueba a todos los niños que nacen en las maternidades (tanto públicas como privadas) de la Comunidad Valenciana, de manera gratuita durante los primeros días de vida.
En caso de que se confirme el diagnóstico de P.K.U. resulta decisivo para que el desarrollo físico y psíquico del niño resulte normal que el tratamiento se instaure lo antes posible.
¿Dónde se tratan estos niños?
El hecho de que se trate de una enfermedad que afecta a pocos niños, hace recomendable que todos ellos sean tratados en el mismo centro, a cargo de especialistas con experiencia en esta enfermedad. En la Comunidad Valenciana, el seguimiento y control de estos niños se realiza en el "Hospital Infantil La Fe".
¿En qué consiste el tratamiento?
El pilar fundamental sobre el que se basa el tratamiento es la dieta, resultando fundamental el control de la calidad de proteínas que se ingieren. Además esta cantidad no es igual para todos los enfermos, sino que debe de ajustarse individualmente. Este tratamiento deberá mantenerse a lo largo de la vida adaptándola a las distintas etapas y circunstancias del desarrollo del niño.
Otras Metabolopatías
También hay otras enfermedades metabólicas de carácter hereditario que, aunque se dan con poca frecuencia, cuando se presentan pueden producir diferentes trastornos muy graves.
Entre estas enfermedades cabe destacar las Aminoacidopatías, Acidurias orgánicas, Enfermedades mitocondriales y Transtornos del Ciclo de la Urea.
Es importante realizar un diagnóstico precoz de estas enfermedades para instaurar un tratamiento precoz, aliviar los síntomas de la enfermedad y realizar un Consejo Genético adecuado.
Si tu hijo se encuentra afectado de Fenilcetonuria o de otras Metabolopatías, en AVAPKU recibirás información, asesoramiento y apoyo de otros padres que ya han vivido la misma experiencia
Existe una Asociación Valenciana de Fenilcetonúricos y otros transtornos Metabólicos(A.V.A.P.K.U.), que ofrece apoyo a todos los enfermos y familiares de estos niños.
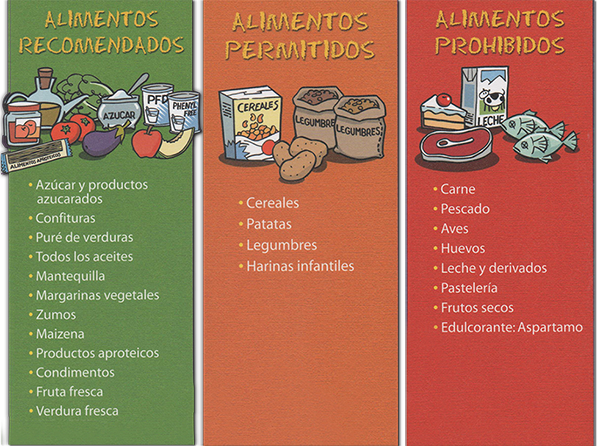
Esquema de alimentos
Alimentos
Esta es una guía de los alimentos que son beneficiosos para la salud del paciente y de los que no lo son.
No debemos olvidar que las dietas son personalizadas para cada paciente en concreto.
Su médico especialista es quien mejor le puede aconsejar en el cuidado y alimentación de su hijo
Guía se la Fenilcetonuria (PKU) para profesores
Es muy importante que los profesores esten bien informados sobre esta enfermedad.
A los niños con PKU hay que tratarlos exactamente igual que a otros niños de la clase. Recuerda! No son diferentes al resto en términos sociales, emocionales, físicos ni mucho menos en su desarrollo académico.
Es importante que se establezca una buena línea de comunicación entre los profesores y los padres a la hora de controlar su alimentación. Los papás de niños con PKU son verdaderos expertos en su alimentación, y deben ser informados.
En el siguiente enlace Guía PKU encontrarán información relacionada con la enfermedad para que esten al día y puedan cuidar mejor de nuestros hijos.
En este otro enlace
 Fichas para colegios.
Fichas para colegios.
Edulcorantes
Los pacientes con PKU no deben tomar aspartamo o lo que es lo mismo edulcorante E-951 y E-162
Adjuntamos un listado donde se indica claramente los edulcorantes PROHIBIDOS y los Si permitidos:
 Edulcorantes prohibidos
Edulcorantes prohibidos
- 951(Aspartamo)
- 962
- Nutrasweet
- Equal
- Canderal
 Edulcorantes permitidos
Edulcorantes permitidos
- 950 - Acesulphame
- 952 - Cyclamate
- 953 - Isomalt
- 954 - Saccharin
- 955 - Sucralose
- 956 - Alitame
- 957 - Thaumatin
- 961 - Neotame
- 965 - Maltitol
- 966 - Lactitol
- 967 - Xylitol
- 968 - Erythritol
- Mannitol, Sobitol
Control bioquímico
Los datos obtenidos mediante el control bioquímico de los pacientes aportarán al clínico elementos objetivos del estado del paciente que permitirán adecuar el tratamiento dietético, si es necesario. El control bioquímico de la PKU implica la determinación de:
- Metabolitos marcadores de control metabólico de la PKU, es decir, compuestos anómalos, por exceso o defecto (fenilalanina y tirosina), implicados en la enfermedad.
- Metabolitos marcadores del estado nutricional de los pacientes, implicados en el correcto crecimiento y desarrollo de los mismos, que pueden hallarse alterados por la dieta restrictiva en proteínas naturales.
- a) La monitorización de la concentración sanguínea de fenilalanina y tirosina se realiza mediante determinación de estos aminoácidos en sangre seca impregnada en papel de filtro (ver Normas para la determinación de fenilalanina en sangre seca).
- La dieta restringida en fenilalanina debe adaptarse para cada niño individualmente, según su tolerancia, con objeto de mantener la concentración sanguínea de fenilalanina recomendada según la edad:
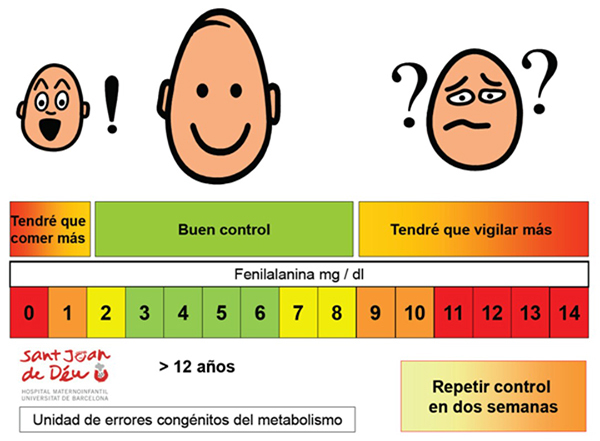
La periodicidad de los controles varía con la edad:
- Menos de 2 meses: semanal.
- De 2 meses a 4 años: quincenal.
- De 4 años en adelante: mensual.
- La punción para obtención de sangre seca se realiza en el domicilio siempre a la misma hora, a ser posible. Las muestras de sangre seca se envían por correo urgente, adjuntando el impreso rellenado convenientemente con los datos actuales del paciente y las incidencias de los últimos días.
- El análisis bioquímico de dichas muestras se realiza semanalmente, informando a las familias del resultado por correo (preferentemente correo electrónico). Cuando la concentración plasmática de fenilalanina no esté dentro de los límites recomendados para la edad y se requieran modificaciones en la dieta, Nutrición contactará con la familia para el ajuste de la misma.
- La evolución de los valores de fenilalanina se representa mediante gráficas que se incluyen en la historia clínica de cada paciente y en las que se puede apreciar fácilmente el control de fenilalanina a lo largo de su vida.
- Además, se calcula el índice de control de la dieta (ICD) como la media de las medianas de los valores de fenilalanina a lo largo de la vida del paciente. El ICD debe ajustarse a los valores recomendados para la edad del paciente.
- b). Los marcadores del estado nutricional de los pacientes con PKU son metabolitos implicados en el correcto crecimiento y desarrollo de los mismos, que pueden hallarse alterados por las dietas restrictivas en determinados nutrientes (ver ejemplo de Control bioquímico anual):
- Vitaminas.
- Oligoelementos.
- Carnitina.
- Ácidos grasos poli-insaturados (PUFAs).
- Marcadores de osteopenia.
Aún cuando la dieta hipoproteica se acompaña con una fórmula especial que contiene todos los aminoácidos excepto fenilalanina y suplementos de vitaminas (A, E, folato, B12, B6), oligoelementos (Fe, Cu, Zn, Se), carnitina, etc, es útil comprobar en los controles bioquímicos periódicos que los pacientes no muestran concentraciones excesivas o defectuosas de alguno de estos compuestos que son indispensables para su desarrollo.
No obstante, el control pobre de la dieta y, sobre todo, el rechazo de las fórmulas especiales en determinados períodos de la vida (adolescencia) pueden determinar las deficiencias nutricionales que se ponen de manifiesto en los controles bioquímicos periódicos.
¿Qué es la fenilcetonuria?
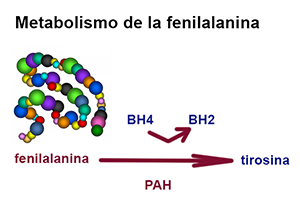
La fenilcetonuria (PKU) es un error congénito del metabolismo, en el que la deficiencia de la enzima fenilalanina hidroxilasa, causa una acumulación de fenilalanina en sangre, orina y tejidos, así como un defecto de tirosina.
¿Qué es la fenilalanina?
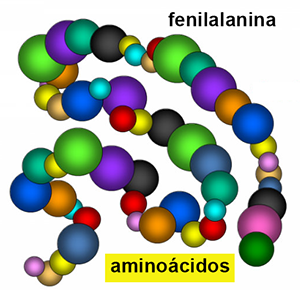
La fenilalanina es un aminoácido, molécula simple que forma parte de las proteínas.
Las proteínas están constituidas por una cadena muy larga de aminoácidos, que se enlazan como las perlas de un collar, en un orden especial para cada una de ellas, que determina su forma en el espacio y con ello, su buen funcionamiento.
Cuando las proteínas se degradan, se liberan los aminoácidos y estos pueden utilizarse para formar otras proteínas nuevas de nuestro organismo o bien para generar otras sustancias y energía.
La fenilalanina tiene su propia vía metabólica, por la cual es capaz de formar un aminoácido muy parecido a ella, la tirosina, gracias a la acción de una enzima, la fenilalanina hidroxilasa (PAH) y de un coenzima que facilita la reacción, la tetrahidrobiopterina (BH4).
Tratamiento de la PKU

Restricción de fenilalanina
El tratamiento clásico de la PKU , descrito por Bickel en 1953, es la restricción de la fenilalanina de la dieta. Esto evita que se acumule este aminoácido en sangre, orina y tejidos y pueda dañar especialmente al cerebro.
Es importante también la suplementación con tirosina, que se halla deficiente debido a la interrupción de la vía metabólica causada por el defecto de PAH.
La tirosina forma parte de las proteínas y es un aminoácido precursor de neurotransmisores.
¿Cómo se realiza la restricción de la fenilalanina de la dieta?
La dieta de los niños con PKU se basa en la reducción del aporte de alimentos ricos en proteínas (por tanto, ricos en fenilalanina).
¿Qué alimentos contienen fenilalanina?
La mayoría de alimentos contienen proteínas en diversa cantidad y por tanto fenilalanina, aunque existen unos más ricos que otros.

- Los alimentos con proteínas de alto valor biológico: carnes, pescados, huevos, leche y derivados (excepto mantequilla) son los que más contienen y están muy restringidos.
- Los alimentos con proteínas de mediano valor biológico: los cereales, legumbres y sus derivados, deben ser controlados en su ingesta.
- Los alimentos con proteínas de bajo valor biológico: las verduras, hortalizas y frutas tienen menor contenido en fenilalanina y su consumo puede ser mayor.
- Alimentos como los aceites y grasas, o bien como el azúcar y la miel, no contienen fenilalanina. El almidón de maíz (maicena) y el almidón de la mandioca (tapioca) contienen cantidades despreciables de fenilalanina.
¿La restricción de proteínas, no causa deficiencias nutricionales?
La restricción proteica debe combinarse con la administración de una fórmula especial sin fenilalanina , que contiene todos los demás aminoácidos, especialmente tirosina, para evitar su deficiencia.
Esta fórmula contiene además vitaminas, oligoelementos y sustancias antioxidantes , para evitar la deficiencia de micronutrientes que podría causar la restricción de las proteínas naturales de la dieta.
¿Se puede suprimir del todo o es necesaria la fenilalanina de la dieta?
La fenilalanina es un aminoácido esencial y es imprescindible para la síntesis de proteínas. Estas son necesarias no sólo para el crecimiento del niño , sino también para que funcione correctamente su metabolismo.
Se necesitan para ello unas concentraciones adecuadas de fenilalanina que se logran con un aporte suficiente a través de la dieta, dependiendo su ajuste final del grado de tolerancia de cada paciente.
¿Cómo se alimentan los niños PKU en la lactancia?
Inicialmente, mientras se alimentan exclusivamente con leche, se retira la lactancia materna o fórmula adaptada si es necesario, sustituyéndola por la fórmula especial sin fenilalanina, hasta que la concentración sanguínea de este aminoácido se halle dentro del rango de valores recomendado
Una vez superada esta fase, se mezclan la fórmula especial exenta de fenilalanina con fórmulas adaptadas o con leche materna, según la tolerancia del paciente, que viene reflejada por la concentración plasmática de fenilalanina.
¿Cómo es la dieta en la infancia?
Pasados los seis primeros meses (lactancia), el niño debe ir incorporando a su dieta la alimentación complementaria.
Además de leche, los niños se alimentan con cereales, purés de verduras o alimentos sólidos, normalmente se disminuye la ingesta de leche adaptada o materna, pero continúan con la fórmula exenta de fenilalanina.
Los principios básicos que orientan la alimentación en el niño PKU son los mismos que en la población general, pero con alimentación adaptada y controlada en proteínas.
Las necesidades calóricas aportadas por los micronutrientes , deben ser las apropiadas para cada edad.
¿Cuánto tiempo deben tomarse las fórmulas exentas de fenilalanina?
Las fórmulas exentas de fenilalanina son imprescindibles durante toda la vida , ya que con una dieta restrictiva en fenilalanina nunca alcanzaríamos a cubrir las necesidades proteicas del individuo.
Además, estas fórmulas contienen todos los micronutrientes (vitaminas y oligoelementos) que normalmente se ingieren con las proteínas naturales, con lo que se evitan los estados carenciales.
¿Cuántas veces al día debe tomarse la fórmula especial?
Es necesario repartir la fórmula en cuatro comidas al día , mínimo tres, para asegurar que la proporción de todos los aminoácidos sea regular a lo largo del día.
¿Hasta qué edad debe mantenerse la dieta restringida en fenilalanina?
La dieta restringida en fenilalanina debe mantenerse durante toda la vida.
Se ha demostrado que la interrupción del tratamiento conduce en algunos pacientes a una disminución en el coeficiente intelectual, con problemas de carácter y comportamiento en forma de agitación, trastornos del sueño, temblores y otras complicaciones neurológicas más graves en algunos casos.
Por otra parte, se ha demostrado también la importancia del buen control dietético en el pronóstico de la enfermedad.
Enlaces
Donaciones

Agradecemos en nombre de la asociación cualquier aportación que podáis hacer; Nº de cuenta:
ES34 2100 8702 5413 0019 1472
Webs amigas







